Jackson Ford, Jie Lu, Charles Liotta and Charles Eckert, “Solvent effects on the kinetics of a Diels-Alder reaction in gas-expanded liquids,” I&EC Res, 47, 632-637, 2008.
Gas-expanded liquids (GXLs) form a unique class of environmentally benign solvents that offer many of the benefits of both organic liquids and supercritical fluids. A more complete understanding of the interactions between the gas, the organic liquid, and solutes at the molecular level will enable the full exploitation of GXLs. Combining kinetic and solvatochromic studies, we have developed a comprehensive multiparameter approach to add insight into the molecular interactions related to a Diels-Alder reaction in CO2-expanded acetonitrile. We have studied the kinetics of the Diels-Alder reaction of anthracene and 4-phenyl-1,2,4-triazoline-3,5-dione as a function of solvent composition using in situ high-pressure fluorescence spectroscopy. We have also measured the values of the Kamlet-Taft solvatochromic parameters * (dipolarity/polarizibility), (hydrogen bonding acidity), and (hydrogen bonding basicity) in CO2-expanded acetonitrile using in situ high-pressure UV/vis spectroscopy. A linear solvation energy relationship (LSER) describes the reaction rate in terms of the measured solvatochromic parameters. We correlate the reaction rates with the solvatochromic parameters based on the LSER and yield satisfactory consistency with the experimental data.
Lam Phan, Daniel Chiu, David J. Heldebrant, Hillary Huttenhower, Ejae John, Xiaowang Li, Pamela Pollet, Ruiyao Wang, Charles A. Eckert, Charles L. Liotta, and Philip G. Jessop, “Switchable Solvents Consisting of Amidine/Alcohol or Guanidine/Alcohol Mixtures,” I&EC Res, 47, 539-545, 2008.
Liquids that consist of a mixture of an alcohol and either an amidine or a guanidine have been developed to switch from a low-polarity form to a high-polarity ionic liquid upon treatment with CO2 at atmospheric pressure. Treatment with N2 and/or mild heat (50-60ºC) reverses the process. These liquids can be used as switchable solvents to dissolve and then precipitate a solute or to dissolve reagents for a chemical synthesis and then precipitate the product.
Aaron M. Scurto, Elizabeth Newton, Ross R. Weikel, Laura Draucker, Jason Hallett, Charles L. Liotta, Walter Leitner, Charles A. Eckert, “Melting Point Depression of Ionic Liquids with CO2: Measurement and Modeling,” I&EC Res, 47, 493-501, 2008.
Development of ionic liquids for specific tasks is currently being pursued by many researchers as numerous cation/anion combinations are theoretically possible. However, only a small fraction of these combinations melt below 100ºC. Recently, large melting point depressions of several ionic solids with compressed carbon dioxide have been reported. This investigation details the melting point depression of a large number of ionic organic compounds (ionic liquids) with gaseous, liquid, and supercritical CO2. Large and previously unreported depressions were observed for some of the ionic solids. This methodology greatly expands the numbers of compounds and functional groups that can be employed in an ionic liquid/compressed gas system for various applications. Thermodynamic analysis indicates that even small amounts of CO2 can lead to substantial melting point depression, due to its very low melting temperature and negative deviations to Raoult's law.
Nan Jiang, Daniele Vinci, Charles L. Liotta, Charles A. Eckert, and Arthur J. Ragauskas, “Piperylene Sulfone: A Recyclable Dimethyl Sulfoxide Substitute for Copper-Catalyzed Aerobic Alcohol Oxidation”, I&EC Res, 47, 627-631, 2008.
Piperylene sulfone was investigated as a recyclable dimethyl sulfide (DMSO) substitute for copper-catalyzed room temperature aerobic alcohol oxidation. Under the optimal conditions, various primary alcohols could be selectively converted into aldehydes in excellent yields with exceptionally high turnover frequency (over 31 h-1). The catalytic system also shows good compatibility with heteroatom-containing (S and N) substrates. Most importantly, the new developed catalytic system could also be recycled and reused for three runs with only slight loss of catalytic activity.
Jason P. Hallett, Pamela Pollet, Charles L. Liotta and Charles A. Eckert, “Reversible In Situ Catalyst Formation,” Accounts of Chemical Research, 41, 458 - 467, 2008.
Acid catalysts play a vital role in the industrial synthesis and production of a plethora of organic chemicals. But, their subsequent neutralization and disposal is also a giant source of waste. For example, for a Friedel−Crafts acylation with AlCl3, a kilogram of product yields up to 20 kg of (contaminated) waste salt. Other processes are even worse, and this waste is both an environmental and economic shortcoming. Here we address this issue by showing a series of acid catalysts where the neutralization is “built in” to the system and thus eliminates waste. Clearly these will not replace all organic and mineral acid catalysts, but they can replace many. Further, we show how these self-neutralizing catalysts can often eliminate unwanted byproducts, improve selectivity, or elimination of mass transfer limitations by changing from heterogeneous to homogeneous systems. They readily facilitate separations and promote recycling, to promote both green chemistry and good economics.
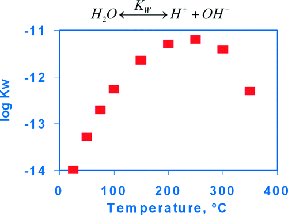
First is near-critical water, or liquid water under pressure, where the KW for dissociation goes up 3−4 decades between 0 °C and 250 °C, thus facilitating both acid and base catalysis. Moreover, as the exothermic hydrogen bonding diminishes, the dielectric constant goes down to the point at which both salts and organics are soluble in this very hot water. For example, toluene and water are completely miscible at 305 °C. This eliminates mass transfer limitations for the reactions, and postreaction cooling not only lowers the KW to neutralize the ions without waste but also results in facile separations from simple liquid−liquid immiscibility.
Further, we show the formation of catalysts with alkylcarbonic acids from alcohols and CO2, analogous to carbonic acid from water and CO2. We show a number of applications for these self-neutralizing catalysts, including the formation of ketals, the formation of diazonium intermediates to couple with electron-rich aromatics to produce dye molecules, and the hydration of β-pinene. Here also these systems often enhance phase behavior to cut mass transfer resistance. In an analogous application we show that peroxide and CO2 gives peroxycarbonic acid, also reversible upon the removal of the CO2, and we show application to epoxidation reactions.
The bottom line is that these catalysts afford profound advantages for both green chemistry and improved economics. The methods outlined here have potential for abundant applications, and we hope that this work will motivate such opportunities.
John L. Gohres, Christopher L. Kitchens, Jason P. Hallett, Alexander V. Popov, Rigoberto Hernandez, Charles L. Liotta, and Charles A. Eckert “A Spectroscopic and Computational Exploration of the Cybotactic Region of Gas-Expanded Liquids: Methanol and Acetone,” J Phys Chem B, 112, 4666-4673, 2008. (abstract)
Local compositions in supercritical and near-critial fluids may differ substantially from bulk compositions, and such differences have important effects on spectroscopic observations, phase equilibria, and chemical kinetics. Here, we compare such determinations around a solute probe dissolved in CO2-expanded methanol and acetone at 25 °C from solvatochromic experiments with molecular dynamics simulations. UV/vis and steady-state fluorescence measurements of the dye Coumarin 153 in the expanded liquid phase indicate preferential solvation in both the S0 and S1 states by the organic species. Simple dielectric continuum models are used to estimate local compositions from the spectroscopic data and are compared to molecular dynamics simulations of a single C153 molecule dissolved in the liquid phase at bubble point conditions. The simulations provide information about the local solvent structure around C153. They suggest the presence of large solvent clustering near the electron-withdrawing side of the probe. Preferential solvation exists in both the S0 and S1 states, but a large disagreement between simulation and experiment exists in the S1 state. Potential reasons for this disparity are discussed.
Jason P. Hallett, Jackson W. Ford, Rebecca S. Jones, Pamela Pollet, Colin A. Thomas, Charles L. Liotta and Charles A. Eckert, “Hydroformylation Catalyst Recycle With Gas-Expanded Liquids,” I&EC Res, 47, 2585-2589, 2008.
Tunable solvents such as gas-expanded liquids offer unique opportunities as benign and economic reaction media. We have improved the turnover frequency of a water-soluble catalyst system by a factor of 85 for the hydroformylation of 1-octene by using a tunable solvent system, which also increases substrate solubility. In our approach, the reaction is run homogeneously in a miscible water-organic solution, and after reaction, CO2 is used as a "miscibility switch" for efficient product and catalyst recovery. Two water-soluble ligands, TPPTS (tris(3-sulfophenyl)phosphine) and TPPMS ((3-sulfophenyl)diphenylphosphine), were compared to triphenylphosphine for hydroformylation activity of their rhodium complexes. The catalyst was recycled three times with no loss of activity.
Jackson W. Ford, Malina E. Janakat, Jie Lu, Charles L. Liotta, Charles A. Eckert, “Local polarity in CO2-expanded acetonitrile: a nucleophilic substitution reaction and 8solvatochromic probes,” J. Org Chem, 73, 3364-3368, 2008.
Many processes that use highly tunable gas-expanded liquids (GXLs) rely on the fact that CO2 addition can greatly affect the polarity of the solvent. We have examined several measures of bulk and local polarity in CO2-expanded acetonitrile to enable more effective exploitation of these polarity changes. The rate of the nucleophilic substitution reaction of tributylamine with methyl p-nitrobenzenesulfonate has been analyzed as a function of solvent composition by using in situ high-pressure UV/vis spectroscopy. We have also measured solvatochromic properties including the Kamlet−Taft π* parameter and Kosower’s Z-value. We correlate these local polarity-based kinetic and solvatochromic measures to develop a better understanding of these property changes as a function of bulk and local solvent composition. The data suggest that local composition enhancement in CO2-expanded acetonitrile has a significant impact on the reaction kinetics.
John L. Gohres, Charu L. Shukla, Rigoberto Hernandez, Charles L. Liotta, and Charles A. Eckert, “Effects of Solute Structure on Local Solvation and Solvent Interactions: Results from UV/Vis Spectroscopy and Molecular Dynamics Simulations,” J Phys Chem B, 112, 14993-14998, 2008.
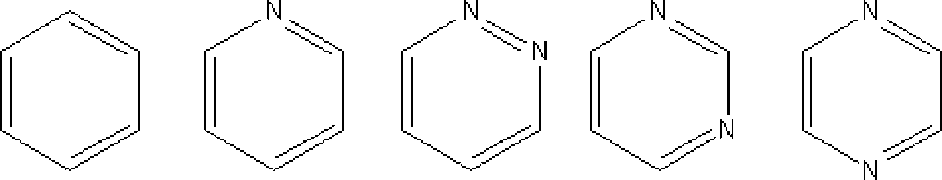
Solvation of heterocyclic amines in CO2-expanded methanol (MeOH) has been explored with UV/vis spectroscopy and molecular dynamics (MD) simulations. A synergistic study of experiments and simulations allows exploration of solute and solvent effects on solvation and the molecular interactions that affect absorption. MeOH−nitrogen hydrogen bonds hinder the n−π* transition; however, CO2 addition causes a blue shift relative to MeOH because of Lewis acid/base interactions with nitrogen. Effects of solute structure are considered, and very different absorption spectra are obtained as nitrogen positions change. MD simulations provide detailed solvent clustering behavior around the solute molecules and show that the local solvent environment and ultimately the spectra are sensitive to the solute structure. This work demonstrates the importance of atomic-level information in determining the structure−property relationships between solute structure, local salvation, and solvatochromism.
|
